
18/10/2020
Ģenētiskie traucējumi – 1.daļa
Esam aizsākuši rakstu sēriju, kurā pavisam nedaudz iepazīstināsim ar dažādām slimībām, kas saistītas ar ģenētiku. Šādas slimības ir pārmantotas vai rodas gēnu mutāciju rezultātā dzīves laikā. Tās var būt gan bīstamas un smagas, gan tādas, kas dzīves kvalitāti īpaši neietekmē. Šādas slimības ir gana daudz, tāpēc sekojiet līdzi mūsu jaunumiem! Šāda veida raksti noteikti būs vēl. Lasi ģenētisko traucējumu pirmo daļu te.
SIA GenEra sāk rakstu sēriju, kurā dosim nelielu ieskatu par dažādām slimībām, kuras rodas ģenētisku traucējumu rezultātā. Visbiežāk slimības raisa izmaiņas kādā DNS posma daļā – bojātu hromosomu vai gēnu mutāciju dēļ. Arī dažādi vides faktori ietekmē ģenētisko izmaiņu rašanos. Dažas no šīm slimībām ir pārmantotas, un mēs tās iegūstam, pārmantojot no vecākiem. Citas, piemēram, audzēji, var izveidoties dzīves laikā, notiekot gēnu mutācijām.
Pirmajā rakstā nedaudz sīkāk pastāstīsim par ģenētiskajām slimībām, kas saistītas ar asins recēšanu, dzelzs uzkrāšanos un holesterīnu. Vairākas no šīm slimībām piedāvājam testēt arī mūsu laboratotrijā, veicot DNS analīzes.
Trombofīlija
FV Leidenes faktora trombofīlija ir iedzimti asins recēšanas traucējumi. Cilvēkiem ar šo mutāciju ir paaugstināta iespējamība asinsvadu aizsprostošanai un dažādu trombožu izveidošanai dzīves laikā. Eiropas populācijā šī mutācija ir diez gan izplatīta, apmēram 3-8% populācijas nēsās savā genomā vismaz vienu mutācijas kopiju.
Simptomi mēdz būt dažādi – ir indivīdi, kas ar šo mutāciju tā arī ar trombofīliju nesaslimst, un ir arī tādi, kuriem tromboze izveidojas jau līdz 30 gadu vecumam. Cilvēkiem ar mutāciju var būt dažādas venozās asinsrites problēmas agrā vecumā, grūtniecības laikā, trombozes var veidoties netipiskās vietās (ne tikai kājās, kas ir tipiski trombozēm), kā arī šādi simptomi var būt ģimenes anamnēzē. Ir dažādi faktori, kas iespējamību saslimt var pastiprintāt – hormonu un kontracepcijas tablešu lietošana, vides faktori, mutāciju skaits (viena vai divas gēna kopijas <- mājaslapā pie trombofīlijas ir mans sacerējums par risku pastip[rinošiem faktoriem) u.c.
Trombofīlija parasti tiek ārstēta ar antikoagulantiem – vielām, kas kavē asins recēšanu. Ja cilvēkam līdz šim tromboze iepriekš nav attīstījusies, tad iesaka izvairīties no citiem faktoriem, kas varētu radīt vēnu aizsprostojumu. Sievietēm ar FV Leidenes mutāciju īpaša uzmanība savai veselībai jāpievērš grūtniecības laikā, jo trombožu risks paaugstinās apmēram septiņas reizes.
Lai noteiktu trombofīlijas gēna mutācijas klātbūtni, tiek taisīts F5 gēna DNS ģenētiskais tests. Šādu analīzi piedāvājam arī GenEra laboratorijā, tā ir daļa no Trombofīlijas riska testa. Sīkāka informācija te.
Hemohromatoze
Iedzimtā hemohromatoze (HH) ir ģenētiska slimība, kas ietekmē dzelzs vielmaiņu organismā. Cilvēkam ar šo slimību dzelzsuzkrājas vairāk kā vajag. Ierasti organisms izlieto dzelzi tik daudz, cik tam nepieciešams, taču hemohromatozes rezultātā šis regulācijas mehānisms ir traucēts. Vairāku gadu laikā liekās dzelzs rezerves uzkrājas aknās, sirdī, aizkuņģa dziedzerī, locītavās un hipofīzē.
Ja slimība tiek laikus atklāta, tad tā labi padodas ārstēšanai, taču, ja kaut kādu iemeslu dēļ tā netiek ārstēta, tā var novest līdz dažādu orgānu bojājumiem un smagām komplikācijām. Slimību ārstē vienkārši – indivīdam regulāri notiek asins noliešana (flebotomija ), lai mazinātu dzelzs līmeni asinīs.
Iedzimto hemohromatozi izraisa mutācijas gēnā HFE. Cilvēkiem ar mantotām abām kļūdainajām gēna kopijām ir lielāka iespējamība slimības attīstībai, taču ne vienmēr tā notiek. Ja mantota ir tikai viena kļūdaina gēna kopija, cilvēks visbiežāk kļūst par slimības nēsātāju ar iespēju bojāto gēnu nodot saviem pēcnācējiem, bet bez klīniskām pazīmēm.
Bieži vien slimības noteikšana ir novēlota, jo simptomi ir līdzīgi daudzām citām slimībām. Ja ir aizdomas par slimību, tiek pārbaudīts transferīns – proteīns, kas asinīs pārvieto dzelzi. Ja tā saturācija ir liela, tas var liecināt par HH. Lai tālāk apstiprinātu vai noraidītu saslimšanu, izmanto DNS testēšanu, meklējot bojātos gēnus. Šādu ģenētisko analīzi veicam arī mūsu, SIA GenEra laboratorijā. Sīkāka informācija par testu meklējama te.
Hiperholesterinēmija
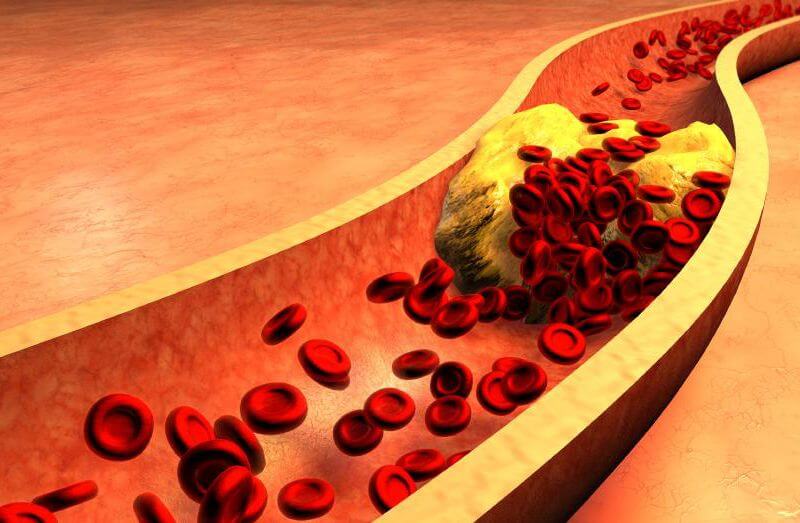
Ģimenes hiperholesterinēmija ir iedzimts stāvoklis, kas izraisa augstu zema bīvuma lipoproteīnu (ZBL) holesterīna līmeni asīnīs jau ļoti agrīnā vecumā (īpaši smagos gadījumos, jau pie dzimšanas). Attiecīgi arī sirdslēkmes šiem pacientiem ir agrīnā vecumā. Holesterīns ir atsevišķs tauku paveids, kas atrodas ķermeņa šūnās. Normāli ķermenim holesterīns ir vajadzīgs, lai sintezētu vitamīnus, palīdzētu vielmaiņā un veidotu hormonus. Taču, ja tā ir par daudz, tas artērijās veido aizsprostojumus un tādejādi paaugstina dažādu sirds slimību risku.
Holesterīns asinīs pārvietojas lipoproteīnu formā. Ir divas galvenās lipoproteīnu formas – zema (saīsinājumā apzīmē ar ZBL) un augsta blīvuma lipoproteīni (ABL). ZBL holesterīns parasti tiek saukts par “slikto holesterīnu”, jo tas visbiežāk rada riskus slimībām. Savukārt ABL holesterīns tiek uzskatīts par “labo”, jo tas palīdz aknām holesterīnu izvadīt.
Tipiskākie ģimenes hip erholesterinēmijas simptomi ir augsts ZBL holesterīna līmenis un tāda pati ģimenes anamnēze, ieskaitot arī sirdslēkmes agrīnos vecumos. Tāpat simptoms var būt stenokardija (sāpes krūtīs), kā arī holesterīna izgulsnējumi dažādās ķermeņa vietās, piemēram, ādā vai cīpslās (ksantomas), plakstiņos (ksantelazmas) vai citur. Ārstēšana parasti tiek mērķēta uz ZBL holesterīna līmeņa samazināšanu gan ar medikamentu, gan uztura maiņas un fizisko aktivitāšu palīdzību. Tiesa iedzimtās, jeb ģimenes hiperholesterinēmijas gadījumā, kad pacientam ir mutācija, kādā no šo slimību izraisošajiem gēniem, ar diētu un fiziskajām aktivitātēm visticamāk nepietiks, lai normalizētu ZBL holesterīna līmeni asinīs.
Viens no galvenajiem gēniem LDLR (Low Density Lipoprotein Receptor jeb Zema Blīvuma Lipoproteīnu Receptors), kas izraisa hiperholesterinēmiju atrodas deviņpadsmitajā hromosomā. Tas satur informāciju par proteīnu - receptoru, kas ir atbildīgs par ZBL izvadīšanu no asinsrites. Ja cilvēks ir viena bojātā gēna nēsātājs jeb heterozigots, tam jau dzīves laikā veidosies dažādi simptomi, kas saistīti ar paaugstinātu ZBL līmeni asinīs. Taču, ja persona manto abus bojātos gēnus, jeb ir homozigota, tad slimības simptomi ir daudz smagāki, tie parādās agrākā vecumā un risks sirds lēkmei un pat nāvei ir jau zem 30 gadu vecuma.
Otrs svarīgs gēns, kura mutācijas ietekmē ģimenes hiperholesterinēmijas atīstību ir APOB (APOlipoproteīs B). Šis gēns kodē proteīnu, kurš atrodas ZBL sastāvā un kuru atpazīst augstāk minētais receptors. Ja šis proteins ir izmainīts (tā kodējošajā gēnā ir kāda mutācija), tad receptors neatpazīst ZBL daļiņu un to nav iespējams aizvākt no asinīm, kā rezultātā ZBL holsterīns uzkrājas asinīs. Šī gēna mutāciju gadījumā, slimniekiem simptomi, parasti ir nedaudz maigāki, jo organismā ir citi papildus mehānismi ZBL holsterīna aizvākšanai no asinīm.
Ģimenes hiperholesterinēmiju diagnosticē pēc asins analīžu rezultātiem, sirds izmeklējumiem, klīniskajiem simptomiem, kā arī ar LDLR un APOB gēnu ģenētisko testēšanu.